Leucemia Linfoblastica Acuta
Definizione
La leucemia linfoblastica acuta (LLA) è una patologia dovuta a alterazioni genetiche a carico dei progenitori dei linfociti, noti come ‘linfoblasti’.1 Queste cellule acquisiscono maggiori capacità di crescita e di sopravvivenza che ne determinano una proliferazione incontrollata nel midollo osseo, nel sangue e in siti extramidollari.2 I linfoblasti si accumulano come precursori immaturi nelle sedi midollari sopprimendo la normale produzione delle cellule ematiche, per cui i pazienti affetti da LLA presentano in genere anemie, neutropenie e trombocitopenie.1
Classificazione
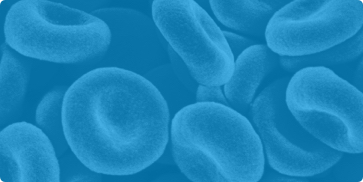
La classificazione attuale dell’Organizzazione Mondiale della Sanità si basa
sulla morfologia e il profilo citogenetico dei linfoblasti. Le due forme
principali sono la LLA-B (LLA a linfociti B) e la LLA-T (LLA a linfociti T).
LLA-B è a sua volta suddivisa in 2 sottotipi: LLA-B con alterazioni genetiche
ricorrenti, in cui si possono identificare specifiche alterazioni
cromosomiche, e LLA-B non altrimenti specificata.2
Classificazione OMS per le forme di leucemia Linfoblastica Acuta (LLA),
revisione 2016
Riprodotto da tabella 1, rif 2
Epidemiologia
L’incidenza di LLA ha una distribuzione bimodale, con il primo picco tra i 2 e i 5 anni, ed un secondo picco verso i 50 anni anche se la maggior parte dei casi di LLA (circa l’80%) si manifestano in età pediatrica.2,3
L’incidenza della LLA in età pediatrica in Italia è di circa 3,5 casi all’anno per 100.000 persone, ed il numero stimato di nuovi casi/anno è di circa 400 mentre l’incidenza nella popolazione adulta è di circa 0,7 casi all’anno per 100.000 persone, con una stima di circa 450 nuovi casi all’anno in Italia.5
Sebbene le strategie di intensificazione della dose abbiano portato a
un miglioramento significativo dei risultati per i pazienti pediatrici, la
prognosi per i soggetti di età più avanzata rimane molto scarsa. Infatti,
nonostante un alto tasso di risposta alla chemioterapia di induzione,
solo il 30-40% dei pazienti adulti con LLA è in grado di ottenere una
remissione a lungo termine.2
Fisiopatologia
La patogenesi della LLA comporta la proliferazione anomala di una popolazione clonale di cellule linfoidi e nella maggior parte dei casi, la LLA si presenta come un neoplasia de novo in individui precedentemente sani.2 Studi sulla
popolazione pediatrica hanno identificato alcune sindromi genetiche come
fattori predisponenti in una minoranza di casi di LLA (sindrome di Down,
anemia di Fanconi, sindrome di Bloom, atassia telangectasia e Sindrome di
rotture cromosomiche tipo Nijmegen).2
Altri fattori
predisponenti comprendono l'esposizione a radiazioni ionizzanti, pesticidi,
alcuni solventi o virus come l’Epstein-Barr o l’HIV.2
La presentazione della LLA può essere aspecifica, con una combinazione di sintomi
costituzionali e segni di insufficienza midollare (anemia, trombocitopenia,
leucopenia). I sintomi più comuni includono i cosiddetti ‘sintomi B’ (febbre,
perdita di peso, sudorazione notturna), facilità all’emorragia o alla comparsa
di lividi, affaticamento, dispnea e infezioni.2
Il coinvolgimento di siti extramidollari si verifica comunemente e può
causare linfoadenopatia, splenomegalia o epatomegalia nel 20% dei
pazienti.
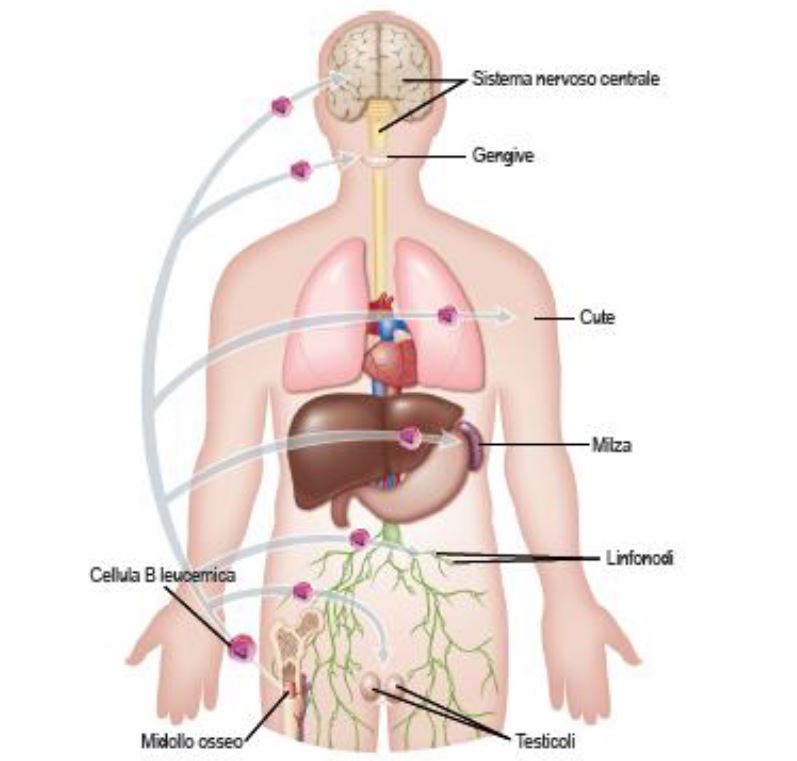
Figura elaborata da testo referenza 4. Sedi comuni di interessamento extramidollare nella
LLA
Il coinvolgimento del Sistema Nervoso Centrale (SNC) al momento della diagnosi si verifica nel 5-8%
dei pazienti
e si presenta più comunemente come deficit dei nervi cranici o meningismo. La
LLA a linfociti T può anche presentarsi con una massa mediastinica.2
Diagnosi
La diagnosi è accertata in presenza di almeno il 20% di linfoblasti nel
midollo osseo o nel sangue periferico. Valutazione morfologica, citometria di
flusso, immunofenotipizzazione e test citogenetici sono utili sia per
confermare la diagnosi sia per la stratificazione del rischio. La puntura
lombare con analisi del liquido cefalo-rachidiano è lo standard
diagnostico per valutare il coinvolgimento del SNC. In caso di esito positivo, deve essere eseguita una risonanza magnetica cerebrale. Altre valutazioni
includono emocromo completo con formula leucocitaria e striscio per valutare
le altre linee cellulari ematopoietiche, i profili di coagulazione e le
chimiche sieriche. L'acido urico basale, il calcio, il fosfato e la lattato
deidrogenasi devono essere rilevati per monitorare la sindrome da lisi
tumorale.2,4
Fattori prognostici e stratificazione del rischio
Una valutazione accurata della prognosi è fondamentale per la gestione della
LLA.2
La stratificazione del rischio consente al medico di
determinare il regime di trattamento più appropriato e quando
considerare il trapianto allogenico di cellule staminali (Allo-SCT).
Storicamente, i criteri utilizzati per la stratificazione del rischio sono stati l'età e la conta leucocitaria al momento della diagnosi.
L'aumento dell'età è un fattore predittivo di prognosi peggiore. I pazienti di
età superiore ai 60 anni hanno prognosi molto scarse, con una sopravvivenza a
lungo termine del 10-15%.2
Sebbene i fattori clinici svolgano
un ruolo importante nella scelta della terapia, le variazioni citogenetiche
hanno un ruolo significativo nella determinazione del rischio. L'aberrazione
citogenetica con il maggiore impatto sulla prognosi e sul trattamento è la
presenza del cromosoma Philadelphia, t (9; 22) che negli adulti rappresenta la mutazione genetica più frequente con una prevalenza dal 10 al 50% che aumenta con l'età.2
Storicamente, la sopravvivenza a
1 anno nei soggetti affetti da LLA Ph-positivi è di circa il 10%.
Inoltre la persistenza di malattia minima residua (MRD) dopo i cicli di induzione o consolidamento rappresenta un altro importante fattore predittivo di prognosi negativa a causa dell’aumentato rischio di recidiva nei pazienti MRD positivi.6
LLA recidivante/refrattaria
Sebbene l'85-90% dei pazienti ottenga la remissione a seguito della terapia di
induzione, vi sono sottogruppi che sono refrattari a questo tipo di terapia.
Inoltre, tra i pazienti che raggiungono una prima remisione completa, circa la metà negli adulti e circa il 15-20% nei pazienti pediatrici successivamente manifestano una recidiva, che determina un considerevole peggioramento della prognosi.7,6 Nei pazienti pediatrici recidivati fattori di stratificazione del rischio sono la sede della recidiva, la durata della prima remissione completa e l’immunofenotipo alla recidiva.6 I pazienti con recidiva precoce (entro i 30 mesi dalla diagnosi), con recidiva isolata nella sola sede midollare e con immunofenotipo T-LLA sono caratterizzati da prognosi generalmente peggiore e perciò considerati ad alto rischio.6
Le opzioni
di terapia di salvataggio per i pazienti R/R Ph-negativa comprendono l’aumento della chemioterapia citotossica e la riformulazione della chemioterapia con un singolo agente.2 Tuttavia, nonostante questo approccio sia in grado di ottenere un modesto effetto di prolungamento della sopravvivenza, la tossicità della terapia citotossica multi-agente può essere limitante e l'unica speranza per la sopravvivenza a lungo termine rimane l’Allo-SCT.2
Tuttavia, recentemente, i nuovi trattamenti immunoterapici
hanno modificato lo scenario della terapia di salvataggio offrendo una
possibilità di cura che potenzialmente possa anche evitare l’effettuazione di
Allo-SCT.2
La malattia minima residua (MRD)
Il termine malattia minima residua (MRD) indica la persistenza di un numero molto ridotto di cellule leucemiche che non è rilevabile dalla citomorfologia convenzionale.7 La MRD viene infatti valutata mediante metodi molecolari e di citometria a flusso estremamente sensibili, almeno 10-4, che permettono di monitorare con maggiore precisione la cinetica della malattia durante e dopo il trattamento.7
Molti studi hanno confermato che la MRD è il fattore prognostico più importante sia nei pazienti pediatrici che negli adulti affetti da LLA, indipendentemente dai tradizionali fattori di rischio.7 Il rilevamento della MRD non è utile solo per la valutazione della risposta iniziale al trattamento e successiva stratificazione del rischio basati sui livelli di MRD, ma anche per monitorare il carico di malattia prima e dopo l’Allo-SCT, per il riconoscimento precoce di recidive e come potenziale endpoint negli studi clinici.7 La MRD viene utilizzata per guidare le decisioni cliniche negli attuali protocolli di trattamento.7